Proteins need to fold
The amino acid sequence determines a protein's folding
Proteins are linear sequences of amino acids; but (with very few exceptions) in order to implement their biological function they need to fold into a well-defined 3-dimensional structure. The ability to do this, and the resulting structure depends crucially on not just the overall amino acid content of the polypeptide, but its specific sequence. In effect, the information required to enable a polypeptide to fold into a protein resides in its specific amino acid sequence. This is known as Anfinsen’s Dogma or the thermodynamic hypothesis.
It is remarkable that each molecule of a particular protein ... has the same precisely defined covalent structure and that when it is in its natural environment, the polypeptides from which it is composed assume the same few conformations even though the complete molecule of the protein is very large. These two properties are foreign to a synthetic chemist. Molecules produced synthetically are either precise but small or large but heterogeneous. Large heterogeneous polymers produced synthetically seldom have defined structures. Yet a molecule of protein is made from atoms held together by the same covalent chemical bonds holding together the smaller molecules to which one is already accustomed. All of the rules of bonding exerted with such unescapability in small molecules are as inescapable in a molecule of protein. [1]
The need for a protein to fold has two important implications:
- it requires the polypeptide to have a minimum length, typically of about 70 amino acids (most are much longer); and
- it imposes stringent constraints on the amino acid sequence.
Before looking further at these we need to say something about biological amino acids.
Amino acids
There are 20 different biological amino acids used in proteins.[2] All but one of them conform to the general pattern shown in Figure 1. It is the nature of the side chain, R, which determines the properties of the specific amino acid, and which is crucial for the structure and functioning of proteins.
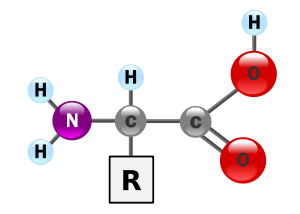
Figure 1. Generalised structure of α-amino acids.[b]
In particular, by virtue of their side chain, eight of the biological amino acids are hydrophobic i.e. ‘oily’, and do not associate readily with water, but may associate readily with each other. They have a particularly important role in protein folding. (Most of the others can be called hydrophilic, i.e. readily associate with water, generally because they are ‘polar’ or electrically charged.)
Hydrophobic effect
The hydrophobic effect is the tendency of nonpolar (‘oily’) substances to aggregate in aqueous solution and exclude water molecules. The reason for the hydrophobic effect is not fully understood and it isn't necessary to enter the debate about this here (but see the note on ’Entropy’).
The important point, so far as protein folding is concerned, is that because the interior of a cell is an aqueous environment, it is energetically favourable to place the hydrophilic amino acids on the outside of the folded protein where they will associate with water, and place hydrophobic ones on the inside where they can associate with each other. It is thought that reducing the number of hydrophobic side chains exposed to water is the principal driving force behind the folding process – so without the hydrophobic amino acids the protein would not fold. The formation of hydrogen bonds and Van der Waals forces within the protein stabilise its structure.
Folding — minimum length of polypeptide
This driving force behind the folding of proteins means not only that without hydrophobic amino acids a polypeptide cannot fold, but imposes a minimum on the length of polypeptide that can fold.
The reason for this is that enough hydrophobic amino acids need to be buried within the core of the folded protein (away from the aqueous cell environment) for the benefits of the hydrophobic effect to be sufficient to overcome the entropic cost of restricting the freedom of movement of all of the polypeptide’s amino acids in going from the unfolded state to a folded protein.
With a very small polypeptide, no matter how tightly it might be folded, none of the amino acids can actually be buried within the folded structure. That is, there is a minimum size before it is possible for a folded protein to have a core which is isolated from the surrounding aqueous environment. But that in itself is not enough.
On average the energetic benefit of burying one hydrophobic amino acid within a folded protein’s core can compensate for the energetic cost of restricting the movement (increased entropy) of 3 amino acids. That is, the hydrophobic core needs to be at least 1/3 of the overall volume of the folded protein in order to have a stable folded structure. Figure 2 illustrates (in 2 dimensions) how the proportion of a folded protein’s core can increase with increasing number of its amino acids.

Figure 2. With increasing size of protein, the higher can be the proportion of buried amino acids.
In turns out that for a polypeptide with a typical mix of amino acids, to be able to form a stable folded structure requires a minimum length of about 70 amino acids.[3]
An interesting illustration of this minimum size for folding comes from the hormone insulin. Insulin comprises two polypeptides of 21 and 30 amino acids (a total of 51) which are covalently joined by two disulphide bridges. If the two polypeptides are obtained separately and then mixed under typical physiological conditions, active insulin does not result – the two polypeptides seem unable to adopt the right configuration. However, insulin is produced biologically via proinsulin which is a single polypeptide of 84 amino acids. Proinsulin spontaneously folds into the correct configuration, the disulphide bridges are formed to stabilise the structure, and the intervening C chain is enzymatically excised to leave biologically active insulin.
Clearly this minimum size has major implications for the origin of new proteins: no longer can it be suggested they might have started off as short polypeptides having only a few amino acids. Whilst there are short polypeptides that don’t fold but have biological activity in the sense of being hormones, transmitters or regulators, no proteins have anything like catalytic activity or a structural role unless they are able to fold into a 3D structure. But that is not all ...
Folding — specificity of the amino acid sequence
It is important to note that it’s no good just having a sufficiently long polypeptide, not even with an adequate proportion of hydrophobic amino acids. In order for the hydrophobic effect to work the hydrophobic amino acids in the core of the protein must be packed together very closely, rather like a 3D jigsaw. An indication of how extraordinarily tightly packed they are can be seen from the percentage of overall volume occupied by atoms: [4]
75% for folded proteins
75% for regularly packed spheres
70-80% for crystals of small organic molecules
∼45% for organic liquids
36% for water
This gives a fair indication of the specificity required – that the amino acid side chains need to be the right shape, size and position so that they can interlock closely, and within the constraints of the polypeptide backbone which links all the amino acids together, the bonds of which cannot rotate freely.
The most important determinant of the final native structure [of a protein] is the steric effect. ... The folding of a polypeptide, however, is a steric nightmare. Not only must all of the functional groups [side chains] fit together in a confined space without overlapping, but all of the functional groups are connected together by the polypeptide. [5]
Degrees of freedom of a polypeptide

Figure 3. Rotations of a polypeptide.[c]
Some covalent chemical bonds can rotate freely, i.e. the groups on either side of the bond could be at almost any angle to each other, like on a swivel. But most are constrained by chemical groups on either side of the bond – which get in the way of each other and prevent free movement.
Figure 3. Rotations of a polypeptide.[c]
Along the backbone of a polypeptide, for each amino acid there are three bonds that potentially can rotate (see Fig. 3):
- the C-N peptide bond between consecutive amino acids, designated ω (omega)
- the C-N bond within an amino acid, designated φ (phi)
- the C-C bond within an amino acid, designated ψ (psi)
However, due to the neighbouring groups:
- the ω bond can adopt only two positions, in fact usually just one;
- the φ and ψ bonds tend to adopt one of three positions.
So the number of positions / conformations each amino acid can adopt is limited to (2 x 3 x 3 =) 18. Clearly this imposes stringent constraints on how the polpeptide can be contorted within the folded protein; and yet proteins are able to do this at the same time as packing the hydrophobic side chains tightly within its core.
If there are only a few conformations that are preferred for each side chain, then there is far less flexibility involved in the folding of a protein than there seems to be at first glance. [6]
This is why the amino acid sequence of most proteins is highly specific, and why most changes (mutations) to a functioning protein’s amino acid sequence, especially those affecting the hydrophobic core, are detrimental, if not completely disruptive.
Of course, evolutionists argue that today’s proteins are the result of many millions of years of evolutionary fine-tuning i.e. that they are now at or near peak performance, which is why further change is detrimental.
However, fine-tuning is one thing; but how did even a crude protein get started in the first place?
- There is nothing in a protein’s amino acid sequence to indicate (even if the organism in question could ‘know’ what to ‘look’ for) whether it will fold or not.
- And natural selection doesn’t help either. For natural selection to work there needs to be at least some function (utility) that can be favoured. But the evidence is that there is no function until the polypeptide is not only long enough but also has a sufficiently closely defined amino acid sequence that it will fold. So a foldable sequence has to be found by chance – there is no step-by-step way to climb this 'mount improbable'.
This is because naturally-occurring polypeptides are created to fold. To fold, they must be composed of hydrophobic and hydrophilic amino acids, placed in a particular sequence, but in a sequence almost impossible to distinguish from a random array.[7]
Currently, even with modern computer processing power, we are unable to predict whether an amino acid sequence will fold – except by comparison with sequences that we know do. (This computational problem is related to Levinthal’s paradox, see below.)
The kinetics of folding — a further constraint?
Cyrus Levinthal (1922-90) realised that even if each amino acid can adopt only 9 positions (accepting that the ω bond generally adopts only one configuration), it suggests that a polypeptide with e.g. 100 amino acids could adopt approximately 9100 ≈ 1095 different conformations. However, because no two (or more) parts of the polypeptide can occupy the same space (referred to as 'excluded volume'), this figure is reduced to about 1050. [8]
Atomic vibrations occur on the scale of femtoseconds, and even if a polypeptide could change to a different conformation every 10-15 second it would take much longer than the age of the universe (~1018 seconds) to try out all of the possible conformations in order to find one that folded (which is why simulations of protein folding are beyond current computer capability).
But most biological proteins fold spontaneously (some require chaperones) in seconds, some in much less than a second.
This is Levinthal's paradox: proteins fold much more quickly than we would expect.[9]
What it means is that there must be something about a polypeptide’s amino acid sequence that is able to direct its folding (which is the subject of considerable research effort). It may be something to do with the resulting folded structure (e.g. it’s secondary structures) that progressively guides the folding process.
However, it may also be that there are some folded states that are theoretically possible but not attainable in practice. That is, in order to be a viable biological protein, not only must it be possible for an amino acid sequence to be able to adopt a folded conformation, but the sequence must be able to provide a route to it – a viable kinetic route (e.g. without localised energetic valleys where the folding process could get stuck).
If this is so, then it imposes a further constraint on the amino acid sequence for a viable biologically active protein.
Folding is not enough
First, to recap, the requirement for a functioning biological protein to fold imposes two essential requirements on its polypeptide:
- A minimum length of about 70 amino acids.
It is misguided wishful thinking to imagine that biologically active proteins could have started off as short amino acid sequences. - Sequence specificity.
To be able to produce a viable protein, a polypeptide must contain not just a suitable mix of hydrophobic and hydrophilic amino acids, but the right ones in the right sequence so that it can be folded into a discrete 3D shape with a very compact hydrophobic core.
In view of these criteria, the requirement to fold – by itself – is probably sufficient to defeat any hope of finding a biologically active protein by opportunistic trial and error.
However, it is important to note that, although these are essential requirements, they are not sufficient to produce a protein with biological activity. That is, a protein that is merely folded – no matter how well – has no biological utility, and will not be favoured by natural selection.
So, as well as – at the same time as – fulfilling the above requirements a potential protein must also incorporate the amino acids – the right ones and in the right places – in order to have a useful function.
For example, an enzyme will require an active site for interacting with its substrate(s). This will require the right amino acids, in the right places in its linear sequence so that – when folded – the appropriate groups from these amino acids will be in the right positions (in 3 dimensions) in relation to each other to e.g. reversibly bind its substrates and/or interact chemically with them. For an example of this, see the requirements for the active site of the enzyme DNA polymerase.
Note – I emphasise – that all of this – the requirements to fold, and e.g. to have a useful active site – must all be met before the protein can offer any utility that could be favoured by natural selection – in order to be just a first step on the route to a fully functioning protein. And, of course, where several proteins are necessarily dependent on each other for their function (which often is the case), then these criteria must be met for all of them – more-or-less together (spatially and temporally) – before any of them can be favoured by natural selection.
We have got to abandon unrealistic woolly notions of how proteins might have arisen, and assess the viability of their evolution in the light of plain biochemical facts, and sober assessment of what natural selection can and cannot do.
Notes
Notes display in the main text when the cursor is on the Note number.
1. Jack Kyte, Structure in Protein Chemistry, 2nd edition, Garland Publishing, 2007, p55.
2. In addition to the usual 20 biological amino acids there are two much less common ones: selenocysteine (where the sulphur atom of cysteine is replaced by selenium) which occurs in most but not all organisms; and pyrrolysine (related to the common amino acid lysine) which occurs only in prokaryotes.
3. Jack Kyte, Structure in Protein Chemistry, 2nd edition, Garland Publishing, 2007, p683.
4. Jack Kyte, Structure in Protein Chemistry, 2nd edition, Garland Publishing, 2007, p278.
5. Jack Kyte, Structure in Protein Chemistry, 2nd edition, Garland Publishing, 2007, p251.
6. Jack Kyte, Structure in Protein Chemistry, 2nd edition, Garland Publishing, 2007, p272.
7. Jack Kyte, Structure in Protein Chemistry, Garland Publishing, 1995, p445.
8. For a polypeptide 100 amino acids long, the effect of excluded volume is to reduce the number of possible configurations by a factor of about 1044; see Ken Dill, Theory for the folding and stability of globular proteins, Biochemistry 24(6), 1501-1509, (1985), p1507.
9. See Wikipedia article 'Levinthal's paradox'.
Image credits
Graphics are by David Swift unless otherwise stated.
Background image for the banner is from https://commons.wikimedia.org/wiki/File:How_proteins_are_made_NSF.jpg and is in the Public Domain.
a. Image from https://commons.wikimedia.org/wiki/File:Christian_B._AnfinsenNIH.jpg; by NIH (https://ihm.nlm.nih.gov/images/B01171) [Public domain], via Wikimedia Commons.
b. Image by GYassineMrabetTalk✉This vector image was created with Inkscape. - Own work, Public Domain, https://commons.wikimedia.org/w/index.php?curid=2551977, from Wikipedia articel 'Amino acid'.
c. Image by Dcrjsr, vectorised Adam Rędzikowski - Own work, CC BY 3.0, https://commons.wikimedia.org/w/index.php?curid=24585750. From Wikipedia article 'Dihedral angle'.
Page created 2017, last revised December 2020.